













Recent Publications

Chemoselective and Diastereoselective Intramolecular (3+2) Cycloadditions of Epoxy and Aziridinyl Enolsilanes
Chen, J. Ling, A. B. Keto, Y. He, K.-H. Low, E. H. Krenske, P Chiu (Selected as a Hot Paper)
Angew. Chem. Int. Ed. 2022, e202116099 doi.org/10.1002/anie.202116099

A soluble iron(ii)-phthalocyanine-catalyzed intramolecular C(sp3)–H amination with alkyl azides
T. You, S.H. Zeng, J. Fan, L. Wu, F. Kang, Y. Liu, C.M. Che
Chem. Commun. 2021,57, 10711 (Selected as Cover)
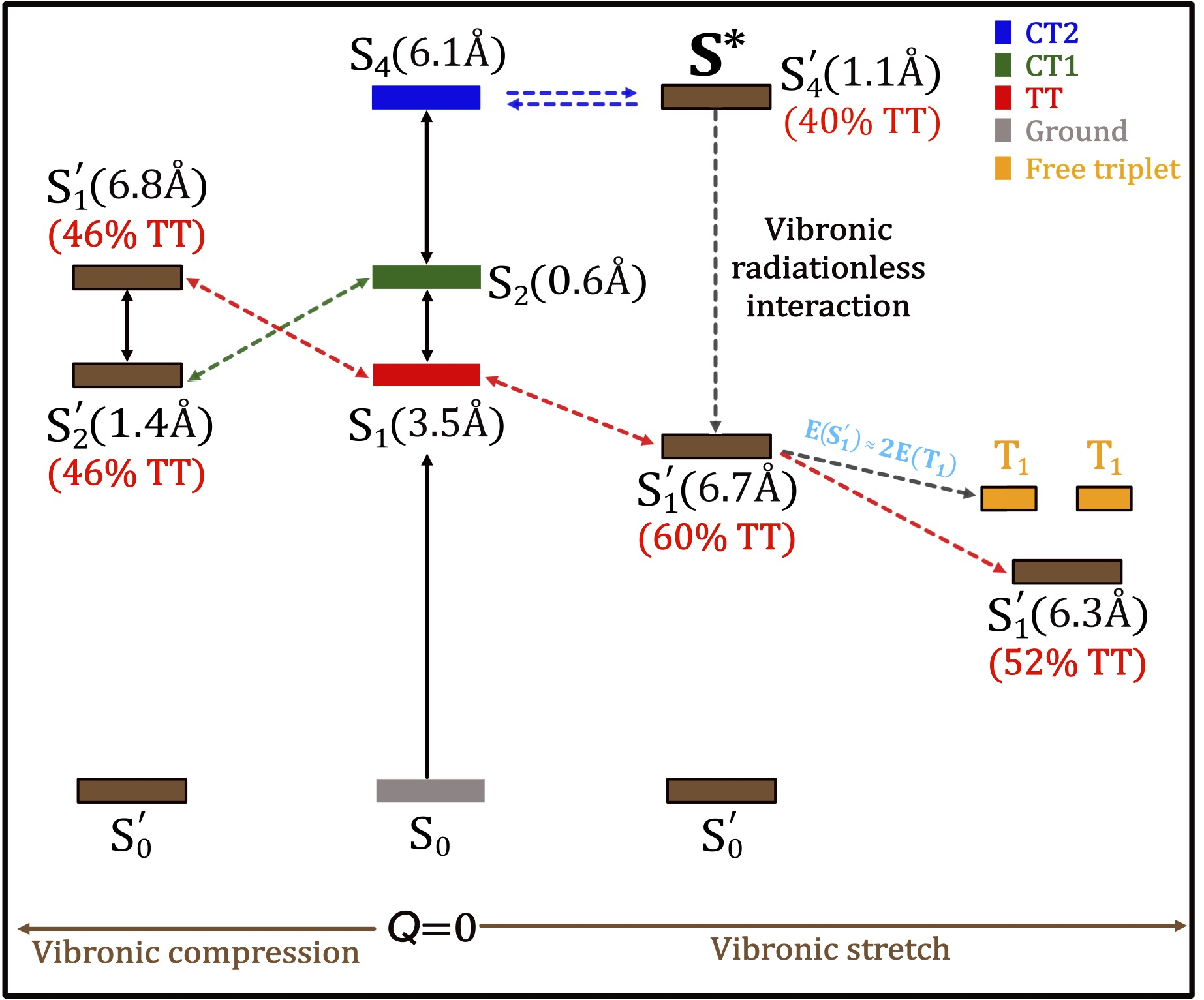
Towards multistate multimode landscapes in singlet fission of pentacene: the dual role of charge-transfer states
R. Walia, Z. Deng, J. Yang
Chem. Sci. 2021 DOI: 10.1039/d1sc01703a
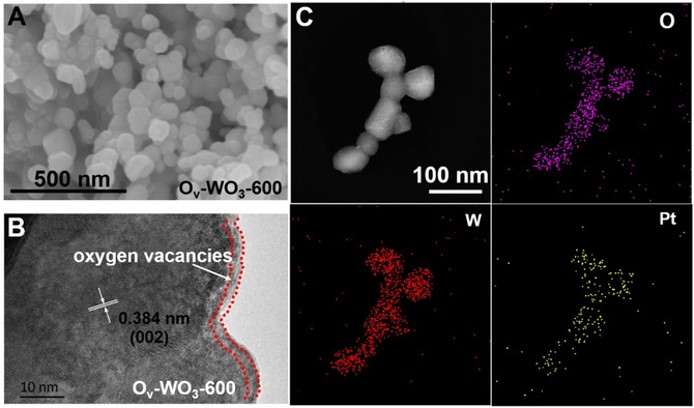
Steering Electron‐Hole Migration Pathways using Oxygen Vacancies in Tungsten Oxides to Enhance their Photocatalytic Oxygen Evolution Performance
Z. Wei, W. Wang, W. Li, X. Bai, J. Zhao, E.C.M. Tse*, D. L. Phillips*, Y. Zhu*
Angew. Chem. Int. Ed. 2021, 60, 8236 DOI:10.1002/anie.202016170
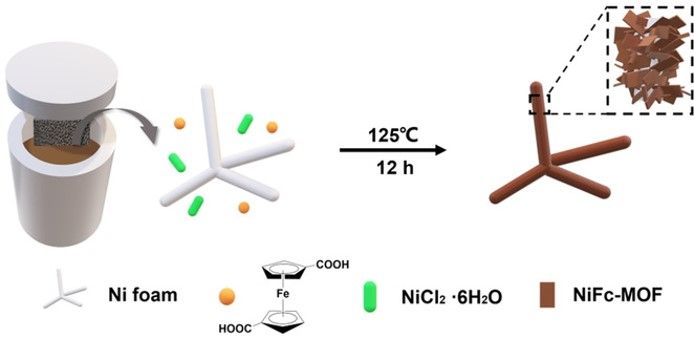
Ferrocene-Based Metal–Organic Framework Nanosheets as a Robust Oxygen Evolution Catalyst
J. Liang, X. Gao, B. Guo, Y. Ding, J. Yan, Z. Guo, E.C.M. Tse*, J. Liu*
Angew. Chem. Int. Ed. 2021, 60, 12770 DOI:10.1002/anie.202101878
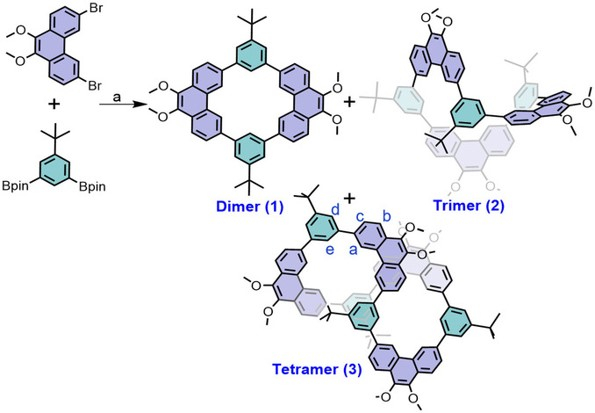
A Molecular Transformer: A π-Conjugated Macrocycle as an Adaptable Host
J. Wang, Y.-Y. Ju, K.-H. Low, Y.-Z. Tan, J. Liu*
Angew. Chem. Int. Ed. 2021, 60, 11814 DOI: 10.1002/anie.202102637
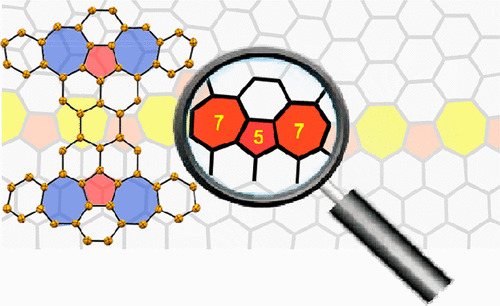
Defective Nanographenes Containing Seven-Five-Seven (7-5-7) Membered Rings
Y. Fei, Y. Fu, X. Bai, L. Du, Z. Li, H. Komber, K.-H. Low, S. Zhou, D. L. Phillips, X. Feng, J. Liu*
J. Am. Chem. Soc. 2021, 143, 2353 DOI: 10.1021/jacs.0c12116
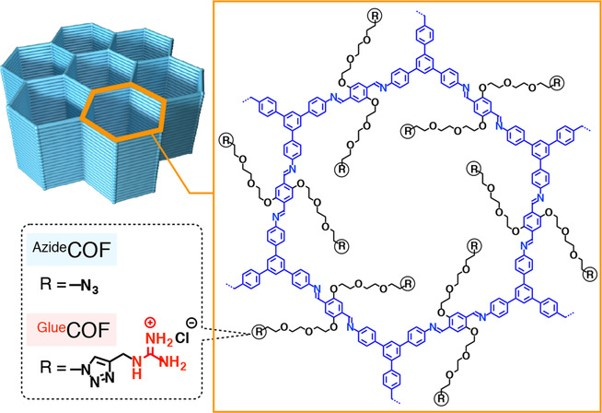
Bio-adhesive Nanoporous Module: Toward Autonomous Gating
H. Jo, T. Kitao, A. Kimura, Y. Itoh, T. Aida*, K. Okuro*
Angew. Chem. Int. Ed. 2021, 60, 8932 DOI: 10.1002/anie.202017117
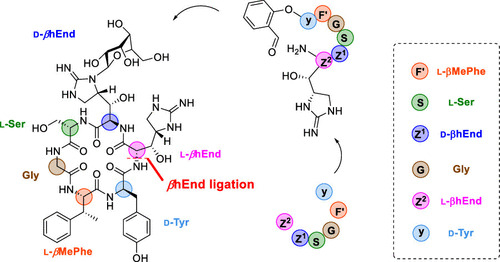
Total synthesis of mannopeptimycin via chemical ligation cyclization
J. Wang, D. Lin, M. Liu, H. Liu, Li, X.*
J. Am. Chem. Soc. 2021. 143, 12784 DOI: 10.1021/jacs.1c05922
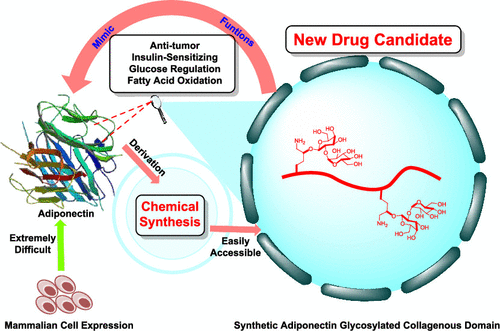
Chemical Synthesis and Biological Evaluation of Adiponectin Collagenous Domain Glycoforms
H. Wu, Y. Zhang, Y. Li, J. Xu, Y. Wang*, X. Li*
J. Am. Chem. Soc. 2021, 143, 7808 DOI: 10.1021/jacs.1c02382
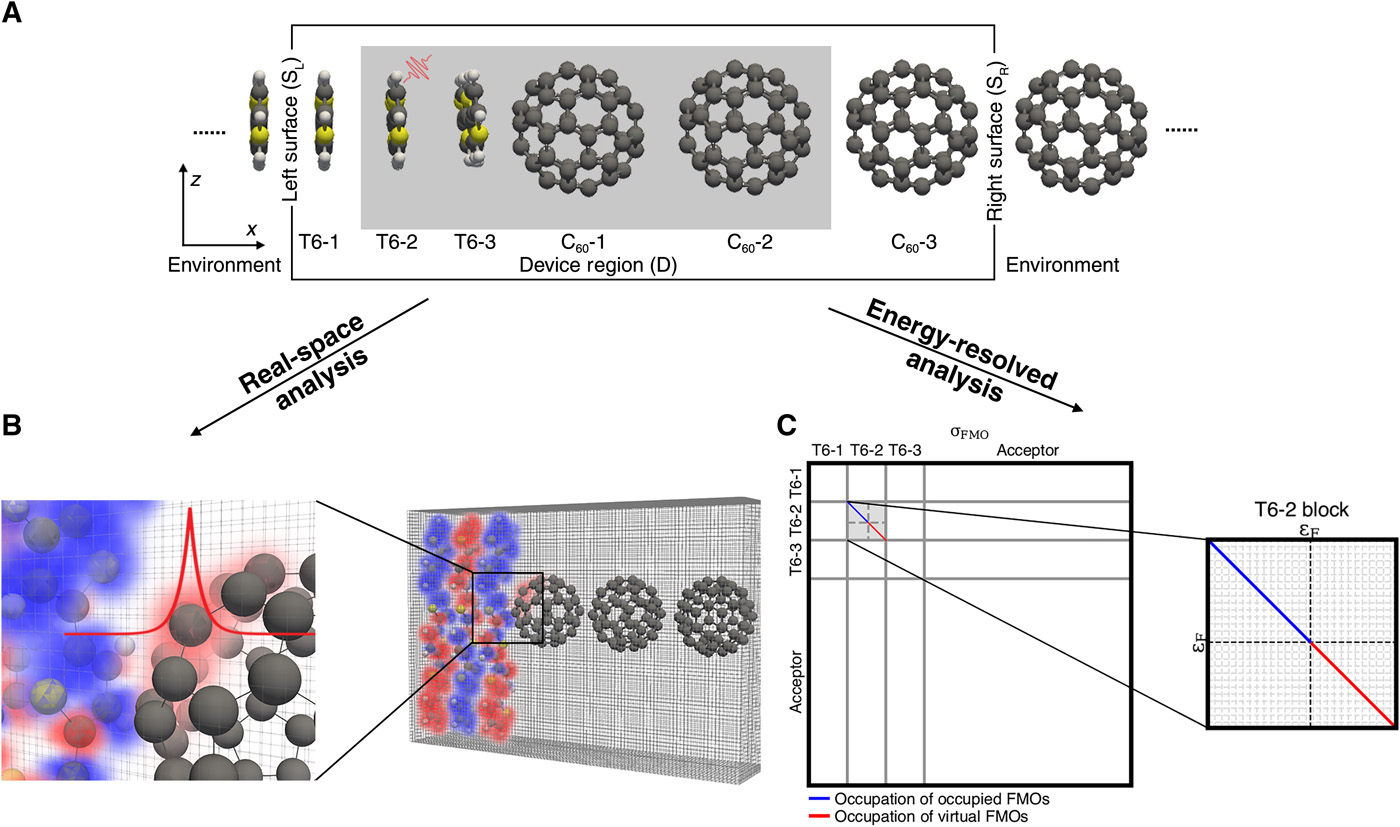
Revealing generation, migration and dissociation of electron−hole pairs and current emergence in an organic photovoltaic cell
Z. Xu, Y. Zhou, C.Y. Yam, T. Frauenheim, C. Lienau, G.H. Chen*
Sci. Adv. 2021, 7, eabf7672 DOI: 10.1126/sciadv.abf7672
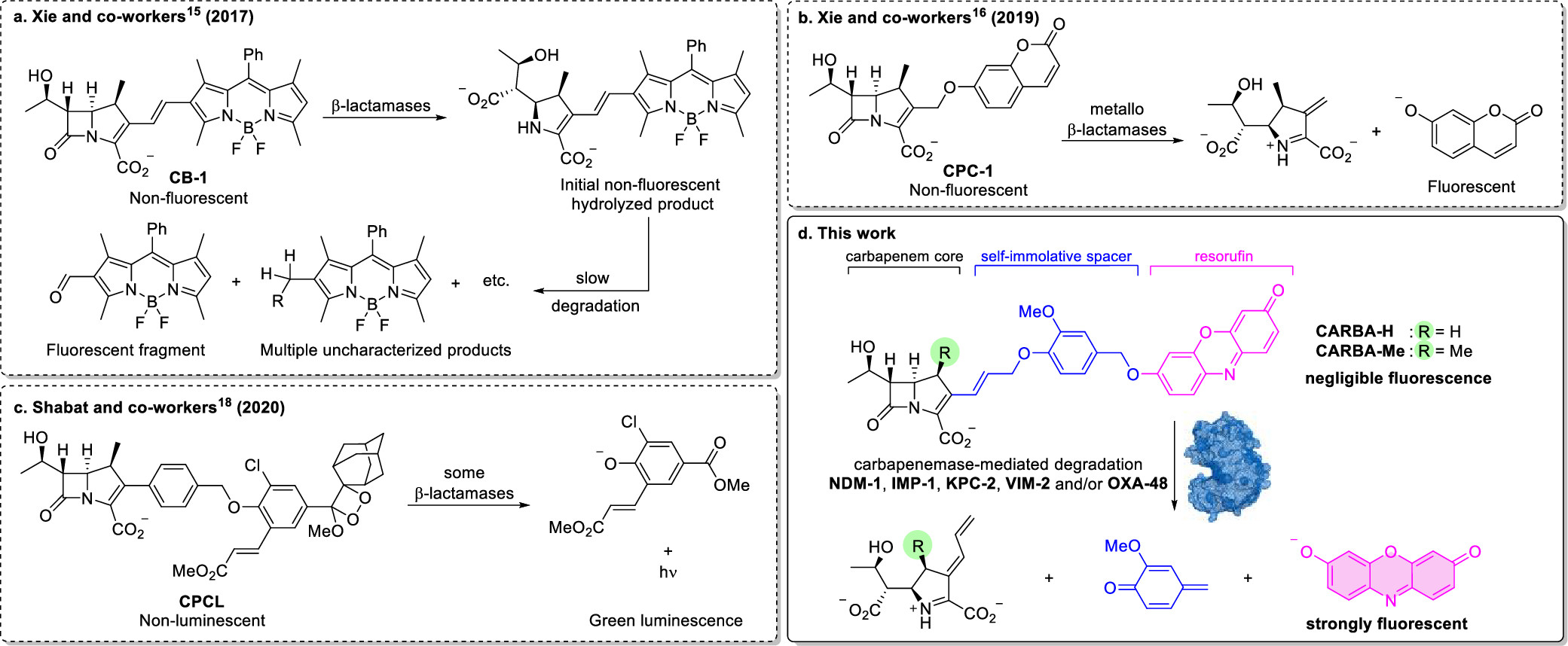
Rapid Broad Spectrum Detection of Carbapenemases with a Dual Fluorogenic-Colorimetric Probe
C.W. Ma, K.K.H. Ng, B.H.C. Yam, P.L. Ho, R.Y.T. Kao, D. Yang*
J. Am. Chem. Soc. 2021, 18, 6886 DOI: 10.1021/jacs.1c00462
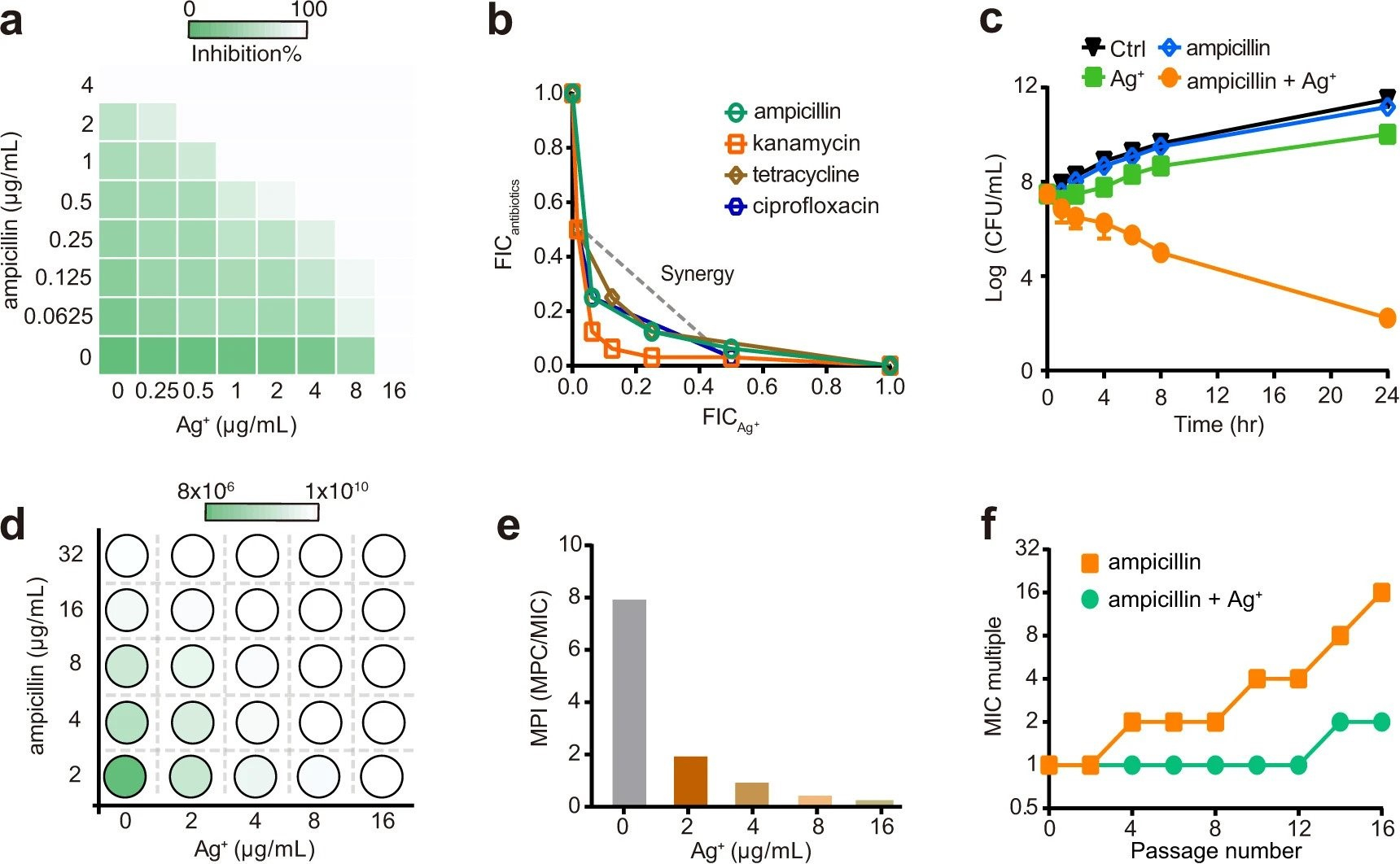
Multi-target mode of action of silver against Staphylococcus aureus endows it with capability to combat antibiotic resistance
H.B. Wang, M.J. Wang, X.H. Xu, P. Gao, Z.L. Xu, Q. Zhang, H.Y. Li, A.X. Yan, R.Y.T. Kao, H.Z. Sun*
Nat. Commun. 2021, 12, 3331 DOI: 10.1038/s41467-021-23659-y
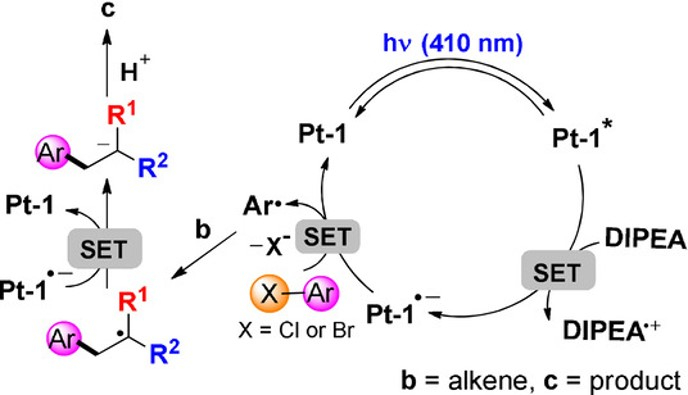
Photoinduced Hydroarylation and Cyclization of Alkenes with Luminescent Platinum(II) Complexes
H. Cheng, T.L. Lam, Y. Liu, Z. Tang, C.M. Che*
Angew.Chem. Int. Ed. 2021, 60, 1383-1389 DOI: 10.1002/anie.202011841
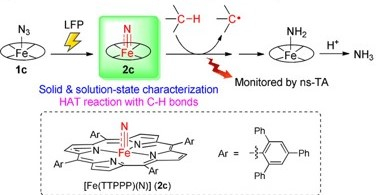
C−H Activation by an Iron-Nitrido Bis-Pocket Porphyrin Species
H.X. Wang, L. Wu, B. Zheng, L. Du, W.P. To, C.H. Ko, D. L. Phillips*, C.M. Che*
Angew.Chem. Int. Ed. 2021, 60,4796 DOI: 10.1002/anie.202014191
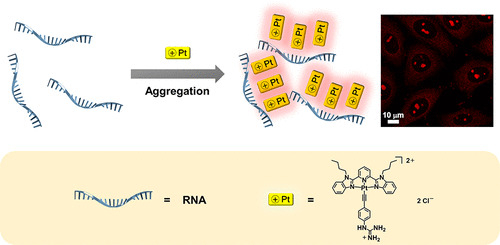
Aggregation and Supramolecular Self-Assembly of Low-Energy Red Luminescent Alkynylplatinum(II) Complexes for RNA Detection, Nucleolus Imaging, and RNA Synthesis Inhibitor Screening
A.S.Y. Law, L.C.C. Lee, K.K.W. Lo*, V.W.W. Yam*
J. Am. Chem. Soc. 2021, 143, 5396 DOI: 10.1021/jacs.0c13327
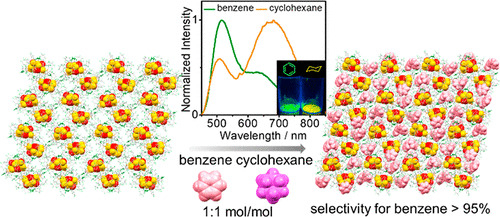
Dual Emissive Gold(I)–Sulfido Cluster Framework Capable of Benzene–Cyclohexane Separation in the Solid State Accompanied by Luminescence Color Changes
L.Y. Yao, V.W.W. Yam*
J. Am. Chem. Soc. 2021, 143, 2558 DOI: 10.1021/jacs.0c11891
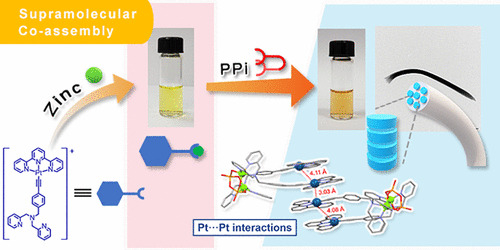
Platinum(II)-Based Host–Guest Coordination-Driven Supramolecular Co-Assembly Assisted by Pt···Pt and π–π Stacking Interactions: A Dual-Selective Luminescence Sensor for Cations and Anions
Y.S. Wong, M. Ng, M.C.L. Yeung, V.W.W. Yam*
J. Am. Chem. Soc. 2021, 143, 973 DOI: 10.1021/jacs.0c11162
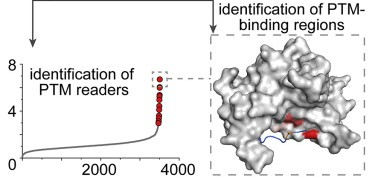
A tri-functional amino acid enables mapping of binding sites for posttranslational-modification-mediated protein-protein interactions
J. Lin, X. Bao*, X.D. Li*
Mol. Cell 2021, 81, 2669 DOI: 10.1016/j.molcel.2021.04.001
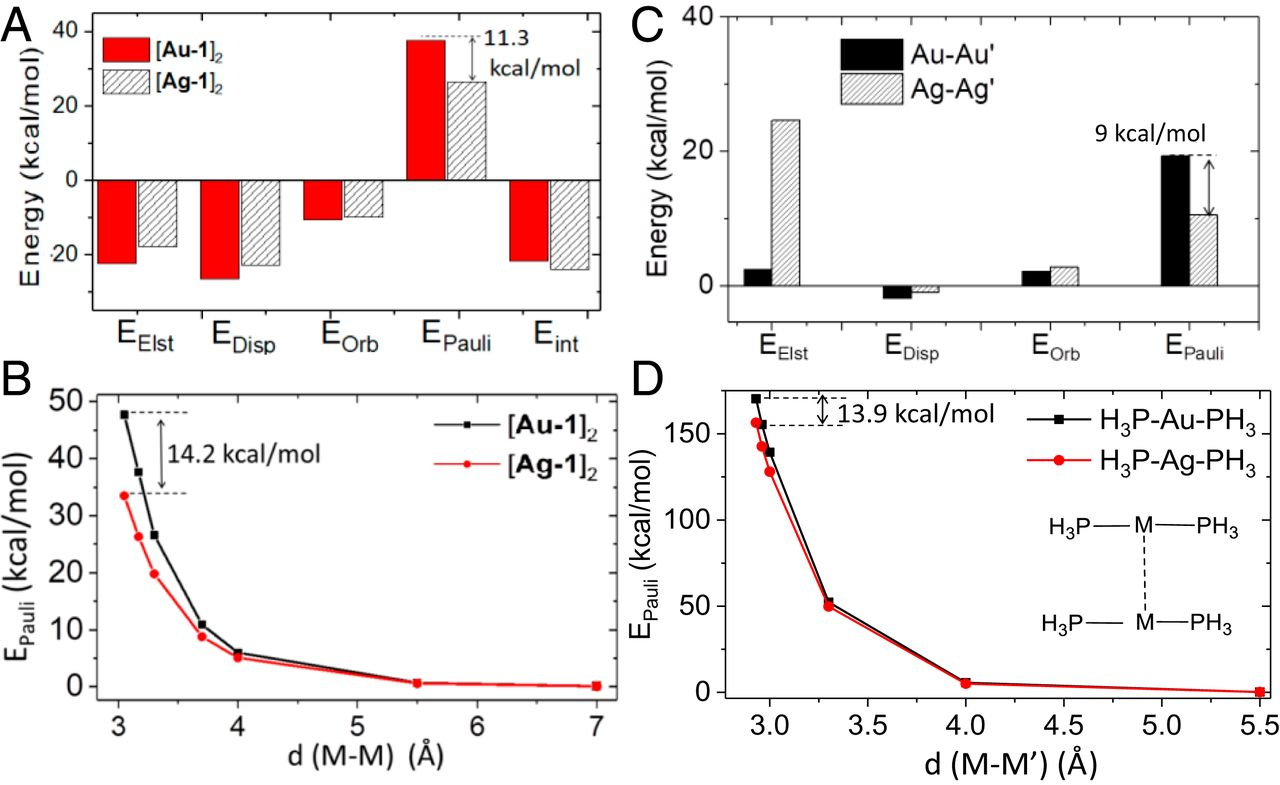
Strong metal–metal Pauli repulsion leads to repulsive metallophilicity in closed-shell d8 and d10 organometallic complexes
Q. Wan*, J. Yang*, W.P. To, C.M Che*
Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2019265118 DOI: 10.1073/pnas.2019265118
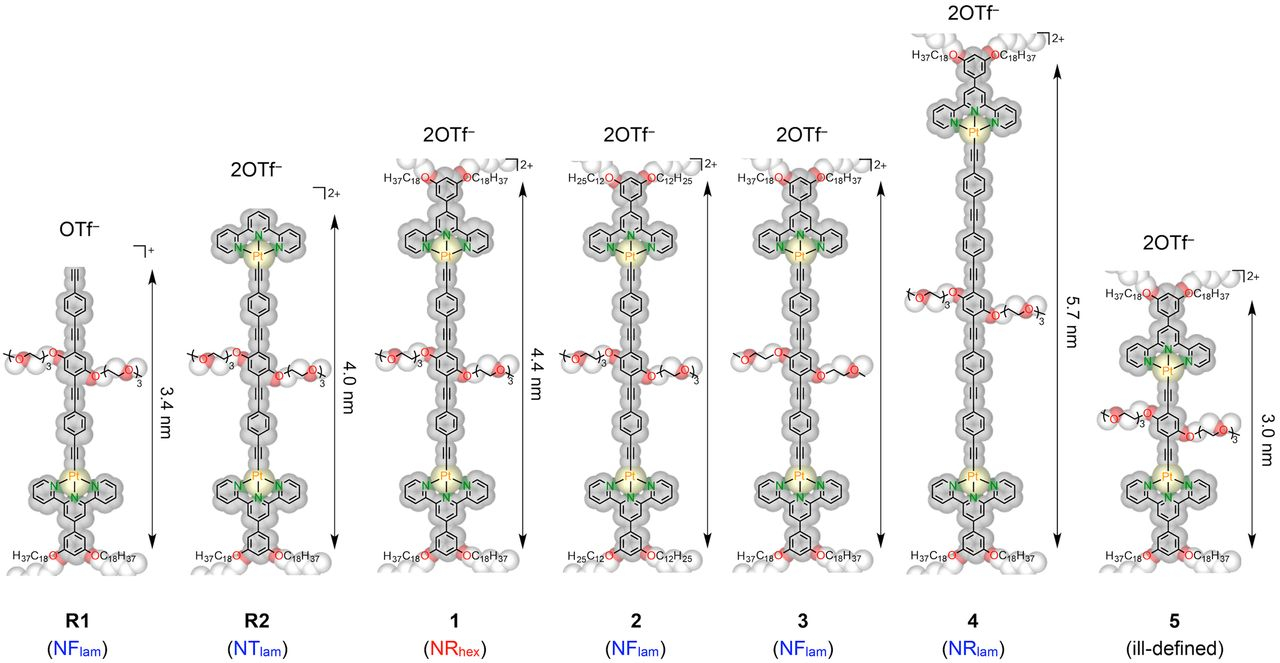
Geometrical manipulation of complex supramolecular tessellations by hierarchical assembly of amphiphilic platinum(II) complexes
J.K.L. Poon, Z. Chen, S.Y.L. Leung, M.Y. Leung, V.W.W. Yam*
Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2022829118
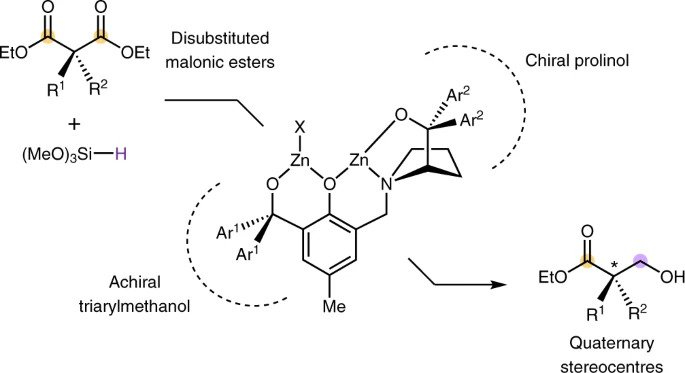
Catalytic reductive desymmetrization of malonic esters
P. Xu, Z. Huang*
Nat. Chem. 2021, 13, 634 DOI: 10.1038/s41557-021-00715-0
Selection of DNA-encoded chemical libraries against endogenous membrane proteins on live cells
Y. Huang, L. Meng, Q. Nie, Y. Zhou, L. Chen, S. Yang, Y. M. E. Fung, X. Li, C. Huang, Y. Cao,* Y. Li,* X. Li*
Nat. Chem. 2021, 13, 77 DOI: 10.1038/s41557-020-00605-x
Department News
- 06 Sep 2023 Nobel Laureate Professor Sir Fraser Stoddart joins HKU as Chair Professor of Chemistry
- 31 Jul 2023 Professor Xuechen LI from HKU's Department of Chemistry Awarded RGC Senior Research Fellow Scheme (SRFS) for 2023/24
- 20 Jul 2023 Biomimetic thermoresponsive superstructures by colloidal soft-and-hard co-assembly
- 07 Jul 2023 Artificial spherical chromatophore nanomicelles for selective CO2 reduction in water
- 07 Jul 2023 Sunlight-powered catalyst transforms methane into valuable chemicals
- 07 Jul 2023 Immobilization of a Molecular Copper Complex and a Carboxylate-Terminated Cocatalyst on a Metal Oxide Electrode for Enhanced Electrocatalytic Oxygen Reduction
Recent Publications
Chemoselective and Diastereoselective Intramolecular (3+2) Cycloadditions of Epoxy and Aziridinyl Enolsilanes
Chen, J. Ling, A. B. Keto, Y. He, K.-H. Low, E. H. Krenske, P Chiu (Selected as a Hot Paper)
Angew. Chem. Int. Ed. 2022, e202116099 doi.org/10.1002/anie.202116099
A soluble iron(ii)-phthalocyanine-catalyzed intramolecular C(sp3)–H amination with alkyl azides
T. You, S.H. Zeng, J. Fan, L. Wu, F. Kang, Y. Liu, C.M. Che
Chem. Commun. 2021,57, 10711 (Selected as Cover)
Towards multistate multimode landscapes in singlet fission of pentacene: the dual role of charge-transfer states
R. Walia, Z. Deng, J. Yang
Chem. Sci. 2021 DOI: 10.1039/d1sc01703a
Steering Electron‐Hole Migration Pathways using Oxygen Vacancies in Tungsten Oxides to Enhance their Photocatalytic Oxygen Evolution Performance
Z. Wei, W. Wang, W. Li, X. Bai, J. Zhao, E.C.M. Tse*, D. L. Phillips*, Y. Zhu*
Angew. Chem. Int. Ed. 2021, 60, 8236 DOI:10.1002/anie.202016170
Ferrocene-Based Metal–Organic Framework Nanosheets as a Robust Oxygen Evolution Catalyst
J. Liang, X. Gao, B. Guo, Y. Ding, J. Yan, Z. Guo, E.C.M. Tse*, J. Liu*
Angew. Chem. Int. Ed. 2021, 60, 12770 DOI:10.1002/anie.202101878
A Molecular Transformer: A π-Conjugated Macrocycle as an Adaptable Host
J. Wang, Y.-Y. Ju, K.-H. Low, Y.-Z. Tan, J. Liu*
Angew. Chem. Int. Ed. 2021, 60, 11814 DOI: 10.1002/anie.202102637
Defective Nanographenes Containing Seven-Five-Seven (7-5-7) Membered Rings
Y. Fei, Y. Fu, X. Bai, L. Du, Z. Li, H. Komber, K.-H. Low, S. Zhou, D. L. Phillips, X. Feng, J. Liu*
J. Am. Chem. Soc. 2021, 143, 2353 DOI: 10.1021/jacs.0c12116
Bio-adhesive Nanoporous Module: Toward Autonomous Gating
H. Jo, T. Kitao, A. Kimura, Y. Itoh, T. Aida*, K. Okuro*
Angew. Chem. Int. Ed. 2021, 60, 8932 DOI: 10.1002/anie.202017117
Total synthesis of mannopeptimycin via chemical ligation cyclization
J. Wang, D. Lin, M. Liu, H. Liu, Li, X.*
J. Am. Chem. Soc. 2021. 143, 12784 DOI: 10.1021/jacs.1c05922
Chemical Synthesis and Biological Evaluation of Adiponectin Collagenous Domain Glycoforms
H. Wu, Y. Zhang, Y. Li, J. Xu, Y. Wang*, X. Li*
J. Am. Chem. Soc. 2021, 143, 7808 DOI: 10.1021/jacs.1c02382
Revealing generation, migration and dissociation of electron−hole pairs and current emergence in an organic photovoltaic cell
Z. Xu, Y. Zhou, C.Y. Yam, T. Frauenheim, C. Lienau, G.H. Chen*
Sci. Adv. 2021, 7, eabf7672 DOI: 10.1126/sciadv.abf7672
Rapid Broad Spectrum Detection of Carbapenemases with a Dual Fluorogenic-Colorimetric Probe
C.W. Ma, K.K.H. Ng, B.H.C. Yam, P.L. Ho, R.Y.T. Kao, D. Yang*
J. Am. Chem. Soc. 2021, 18, 6886 DOI: 10.1021/jacs.1c00462
Multi-target mode of action of silver against Staphylococcus aureus endows it with capability to combat antibiotic resistance
H.B. Wang, M.J. Wang, X.H. Xu, P. Gao, Z.L. Xu, Q. Zhang, H.Y. Li, A.X. Yan, R.Y.T. Kao, H.Z. Sun*
Nat. Commun. 2021, 12, 3331 DOI: 10.1038/s41467-021-23659-y
Photoinduced Hydroarylation and Cyclization of Alkenes with Luminescent Platinum(II) Complexes
H. Cheng, T.L. Lam, Y. Liu, Z. Tang, C.M. Che*
Angew.Chem. Int. Ed. 2021, 60, 1383-1389 DOI: 10.1002/anie.202011841
C−H Activation by an Iron-Nitrido Bis-Pocket Porphyrin Species
H.X. Wang, L. Wu, B. Zheng, L. Du, W.P. To, C.H. Ko, D. L. Phillips*, C.M. Che*
Angew.Chem. Int. Ed. 2021, 60,4796 DOI: 10.1002/anie.202014191
Aggregation and Supramolecular Self-Assembly of Low-Energy Red Luminescent Alkynylplatinum(II) Complexes for RNA Detection, Nucleolus Imaging, and RNA Synthesis Inhibitor Screening
A.S.Y. Law, L.C.C. Lee, K.K.W. Lo*, V.W.W. Yam*
J. Am. Chem. Soc. 2021, 143, 5396 DOI: 10.1021/jacs.0c13327
Dual Emissive Gold(I)–Sulfido Cluster Framework Capable of Benzene–Cyclohexane Separation in the Solid State Accompanied by Luminescence Color Changes
L.Y. Yao, V.W.W. Yam*
J. Am. Chem. Soc. 2021, 143, 2558 DOI: 10.1021/jacs.0c11891
Platinum(II)-Based Host–Guest Coordination-Driven Supramolecular Co-Assembly Assisted by Pt···Pt and π–π Stacking Interactions: A Dual-Selective Luminescence Sensor for Cations and Anions
Y.S. Wong, M. Ng, M.C.L. Yeung, V.W.W. Yam*
J. Am. Chem. Soc. 2021, 143, 973 DOI: 10.1021/jacs.0c11162
A tri-functional amino acid enables mapping of binding sites for posttranslational-modification-mediated protein-protein interactions
J. Lin, X. Bao*, X.D. Li*
Mol. Cell 2021, 81, 2669 DOI: 10.1016/j.molcel.2021.04.001
Strong metal–metal Pauli repulsion leads to repulsive metallophilicity in closed-shell d8 and d10 organometallic complexes
Q. Wan*, J. Yang*, W.P. To, C.M Che*
Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2019265118 DOI: 10.1073/pnas.2019265118
Geometrical manipulation of complex supramolecular tessellations by hierarchical assembly of amphiphilic platinum(II) complexes
J.K.L. Poon, Z. Chen, S.Y.L. Leung, M.Y. Leung, V.W.W. Yam*
Proc. Natl. Acad. Sci. U.S.A. 2021, 118, e2022829118
Catalytic reductive desymmetrization of malonic esters
P. Xu, Z. Huang*
Nat. Chem. 2021, 13, 634 DOI: 10.1038/s41557-021-00715-0
Selection of DNA-encoded chemical libraries against endogenous membrane proteins on live cells
Y. Huang, L. Meng, Q. Nie, Y. Zhou, L. Chen, S. Yang, Y. M. E. Fung, X. Li, C. Huang, Y. Cao,* Y. Li,* X. Li*
Nat. Chem. 2021, 13, 77 DOI: 10.1038/s41557-020-00605-x
Department News
-
06 Sep 2023
Nobel Laureate Professor Sir Fraser Stoddart joins HKU as Chair Professor of Chemistry
-
31 Jul 2023
Professor Xuechen LI from HKU's Department of Chemistry Awarded RGC Senior Research Fellow Scheme (SRFS) for 2023/24
-
20 Jul 2023
Biomimetic thermoresponsive superstructures by colloidal soft-and-hard co-assembly
-
07 Jul 2023
Artificial spherical chromatophore nanomicelles for selective CO2 reduction in water
-
07 Jul 2023
Sunlight-powered catalyst transforms methane into valuable chemicals
-
07 Jul 2023
Immobilization of a Molecular Copper Complex and a Carboxylate-Terminated Cocatalyst on a Metal Oxide Electrode for Enhanced Electrocatalytic Oxygen Reduction
Seminars
-
The 18th Chinese International Peptide Symposium (CPS) 202430Jun2024Department of Chemistry, HKU30 June - 2 July 2024, Hong Kong Ocean Park Marriott Hotel
-
Integrating Chemical Tools to Interrogate Protein Methylation24May2024Prof. Minkui LuoChemical Biology Program, Memorial Sloan Kettering Cancer Center, New York5:00 - 6:00 PM (HKT), CYCP2
-
Photocatalytic Generation of Solar Fuels and Commodity Chemicals22May2024Prof. Francesca ArcudiDepartment of Chemistry Sciences, University of Padova, Italy5:00 - 6:00 PM (HKT), CYCP1